Theoretical Molecular Science Laboratory
Department of Chemistry
Indian Institute of Technology Bombay

Research Area
Our research group focuses on advancing electronic structure theory by integrating quantum computing and classical computational methods to tackle challenges in quantum chemistry

Classical Many-Body Methods:
-
Efficient Coupled Cluster: We develop advanced coupled cluster methods to accurately calculate ground and excited state energies, as well as molecular properties.
-
Machine Learning-Driven Approaches: We leverage machine learning techniques to accelerate and improve the accuracy of classical electronic structure calculations.
-
Large Chemical System Calculations: We employ fragment-based methods to study large chemical systems that are beyond the reach of traditional ab initio methods
-
A Double Exponential Coupled Cluster Theory in the Fragment Molecular Orbital Framework Chakraborty, Anish; Tribedi, Soumi; Maitra, Rahul; J. Chem. Phys. 2022, 156, 244117
-
Formulation of a Dressed Coupled Cluster Method with Implicit Triple Excitations and Benchmark Application to Hydrogen-Bonded Systems Tribedi, Soumi; Chakraborty, Anish; Maitra, Rahul; J. Chem. Theory Comput. 2020, 16, 10, 6317
Dimensionality Reduction for Resource Efficiency:
-
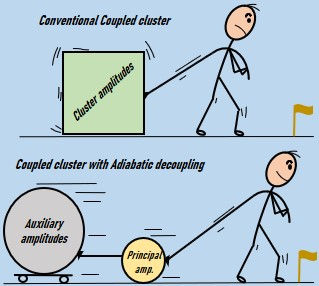
Adiabatic Decoupling: We apply adiabatic decoupling techniques to reduce the dimensionality of the quantum simulation problem, leading to significant computational savings.
-
Feedback-Controlled Optimization: We develop feedback-control strategies to optimize quantum algorithms.
-
A Synergistic Approach towards Optimization of Coupled Cluster Amplitudes by Exploiting Dynamical Hierarchy Patra, Chayan ; Agarawal, Valay; Halder, Dipanjali; Chakraborty, Anish; Mondal, Dibyendu; Halder, Sonaldeep; Maitra, Rahul ChemPhysChem 2022, e202200633
-
An Approximate Coupled Cluster Theory via Nonlinear Dynamics and Synergetics: the Adiabatic Decoupling Conditions Agarawal, Valay; Patra, Chayan; Maitra, Rahul; J. Chem. Phys. 2021, 155, 124115
Chemistry-Inspired Ansatz Design:
-
Ansatz Compactification: We design compact and efficient quantum circuits that capture the essential physics of chemical systems, reducing the number of quantum measurements and gates required.
-
Machine Learning and Generative AI-Driven Approaches: We utilize machine learning and generative AI techniques to automate the design of quantum circuits and to learn optimal parameters for quantum simulations

-
Noise-independent route toward the genesis of a COMPACT ansatz for molecular energetics: A dynamic approach Halder, D; Mondal, D; Maitra, Rahul; J. Chem. Phys. 2024, 160, 124104
-
Machine learning assisted construction of a shallow depth dynamic ansatz for noisy quantum hardware S Halder, A Dey, C Shrikhande, R Maitra; Chem. Sci. 2024, 15, 3279-3289
-
Development of a Compact Ansatz via Operator Commutativity Screening: Digital Quantum Simulation of Molecular Systems Mondal, Dibyendu; Halder, Dipanjali; Halder, Sonaldeep; Maitra, Rahul; J. Chem. Phys. 2023, 159, 014105
Quantum Computing for Electronic Structure:

-
Quantum Error Mitigation: We investigate techniques to reduce the impact of noise and errors in quantum hardware, enabling more reliable and accurate quantum simulations.
-
Hardware-Adaptable Ansatz Design: We develop quantum algorithms that can be efficiently implemented on near-term quantum devices, taking into account the limitations of current hardware.
-
Quantum Phase Estimation: We explore methods for accurately estimating the energy eigenvalues of quantum systems, which is crucial for predicting molecular properties and reaction dynamics.
-
Resource Optimization: We aim to minimize the computational cost of quantum simulations by optimizing the number of measurements and gates required.
-
Development of zero-noise extrapolated projective quantum algorithm for accurate evaluation of molecular energetics in noisy quantum devices S Halder, C Shrikhande, R Maitra; J. Chem. Phys. 2023, 159, 114115
-
Toward a resource-optimized dynamic quantum algorithm via non-iterative auxiliary subspace corrections Patra, C; Mukherjee, D; Halder, S; Mondal, D; Maitra, R; J. Chem. Phys. 2024, 161, 144119
-
Iterative Quantum phase estimation with variationally prepared reference state Halder, Dipanjali; Prasannaa V., Srinivasa; Agarawal, Valay; Maitra, Rahul; Int. J. Quantum Chem. 2022, e27021